

05/06/2026 17:14:55

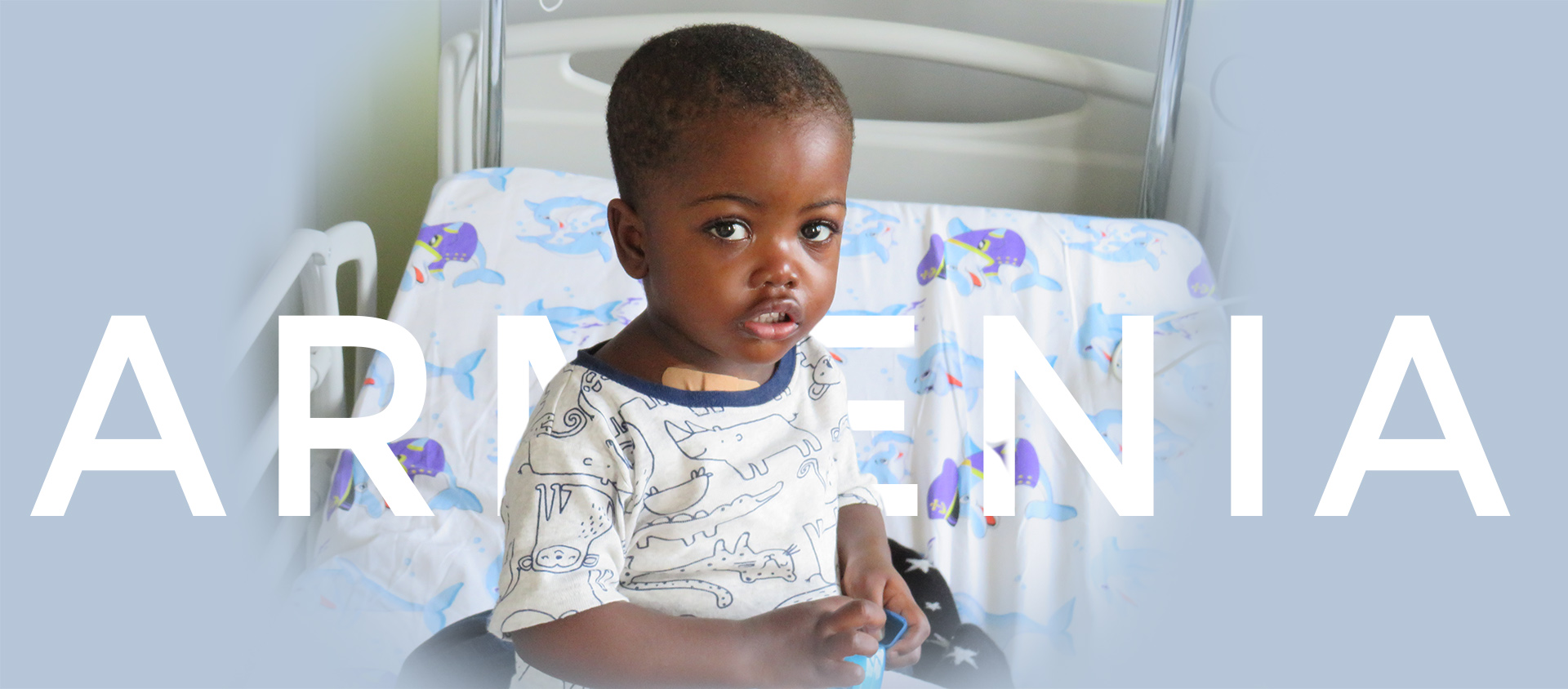
Dal 2007 promuoviamo la cura dei bambini affetti da malattie oncologiche direttamente nel loro Paese. Sosteniamo lo sviluppo sanitario locale con progetti di assistenza avanzata, trasparenti e sostenibili.

Un aiuto che non ti costa nulla... Ma può far tanto.
Per aiutarci, basta scrivere sulla tua denuncia dei redditi il nostro Codice Fiscale
05712190486

Con una tua donazione libera, potrai sostenere i nostri progetti finalizzati alle cure mediche e alla ricerca scientifica. Scopri nella pagina dedicata tutte le modalità per dare un sostegno concreto.
Scopri di più
Possiamo creare insieme tante iniziative solidali: la fantasia è l’unico limite. Scopri come trasformare ogni evento in un’occasione per donare e ottenere vantaggi fiscali.
Scopri di piùAiutaci ad aiutare! Scopri tutte le raccolte fondi attive sul nostro sito.
Nulla è più importante di um bambino... Nulla è più importante di curare e guarire un bambino.
Scopri gli articoli della fondazione Cure2Children! Vedi tutto nella pagina Blog
Per vedere le pubblicazioni scientifiche clicca qui